Evaluation of ANI calculators for microbial genomes
The massive amount of metagenomic sequencing has created a challenge: tons of microbial genomes assembled and binned from environmental samples. To compare those genomes (or MAGs) to investigate genomic diversity, average nucleitide identity (or ANI) is the most widely used and reliable way to calculate genome identity. Alignment-based ANI calculators, e.g., OrthoANI or ani.rb (enveomics package) are too slow for a dozens of genomes.
FastANI or the recent skani represent important breakthroughs of the field in terms of the underlying data structures and algorithms. Minimizer-Jaccard estimator (1), as in MashMap or fastANI (2), as a new data structure for finding homologuous seqeunces in genome pairs, is very efficient and accurate. Skani applies sparse chaining algorithm and can be even faster for high ANI values, but lose accuracy for ANI below 85% or so (3) because essentially, skani skipped computations for ANI below 82% in all versus all comparisons (a majority pair of genomes in GTDB is below 82% ANI, >90%). For above 85% ANI value computations, we can use even faster MinHash algorithms such as One Permutation Hashing with Optimal Densification (a significant improve over Mash, the traditional MinHash using bottom-k implementation) (4), which is at least 100 times faster than skani for running all versus all comparisons for ~300,000 genomes and it does not skip any comparisons. I compare alignment-based ANI, ANIu (orthoANI based on Usearch) with fastANI, skani and One Permutation Hashing with Optimal Densification (our implementation). I found that skani, despite being fast for >85% ANI, has much large variation (e.g., standard deviation) for 85% ANI or below. It is same with One Permutation Hashing with Optimal Densification but skani is much slower.
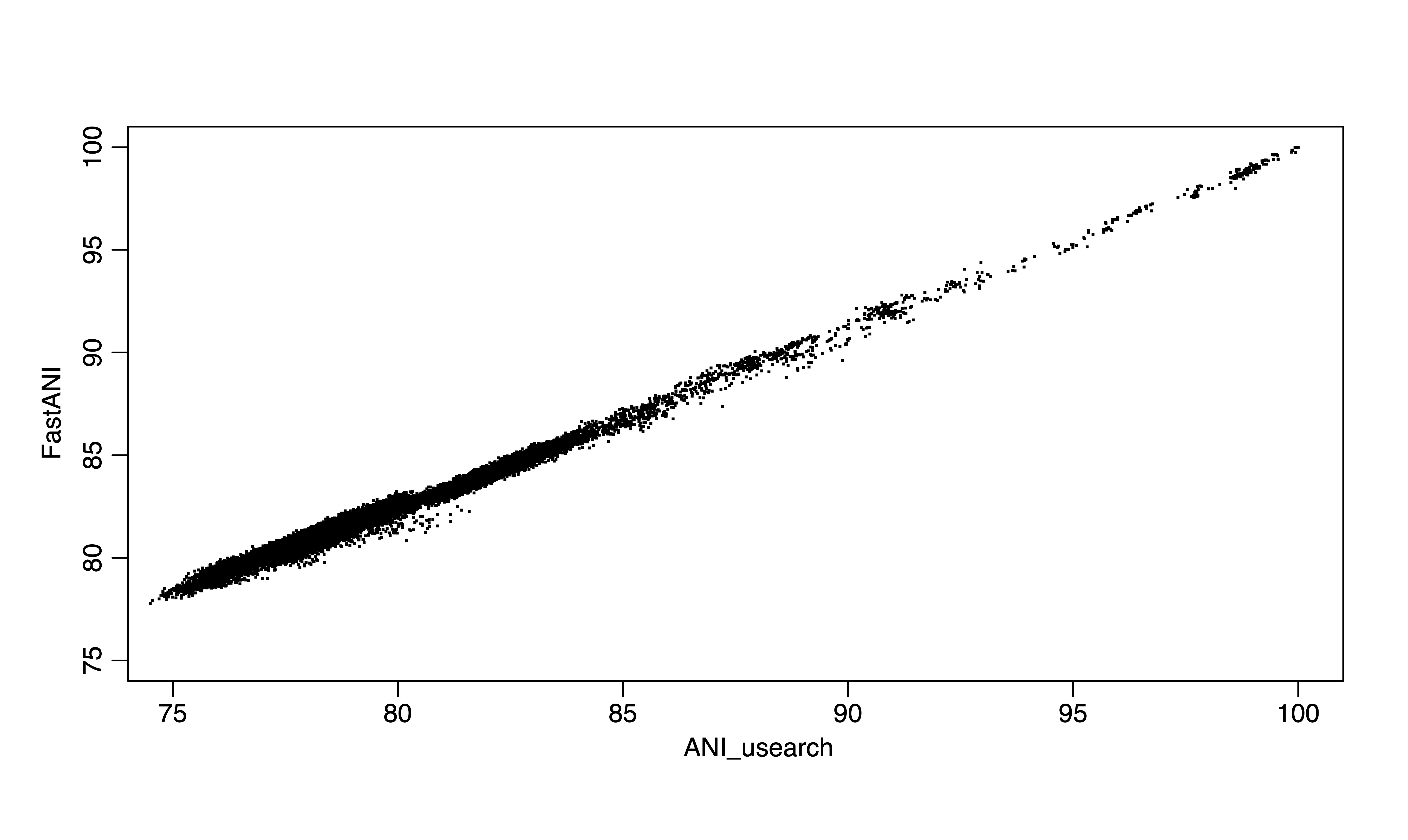 Figure 1. FastANI vs ANIu (usearch) for 300 genomes.
Figure 1. FastANI vs ANIu (usearch) for 300 genomes.
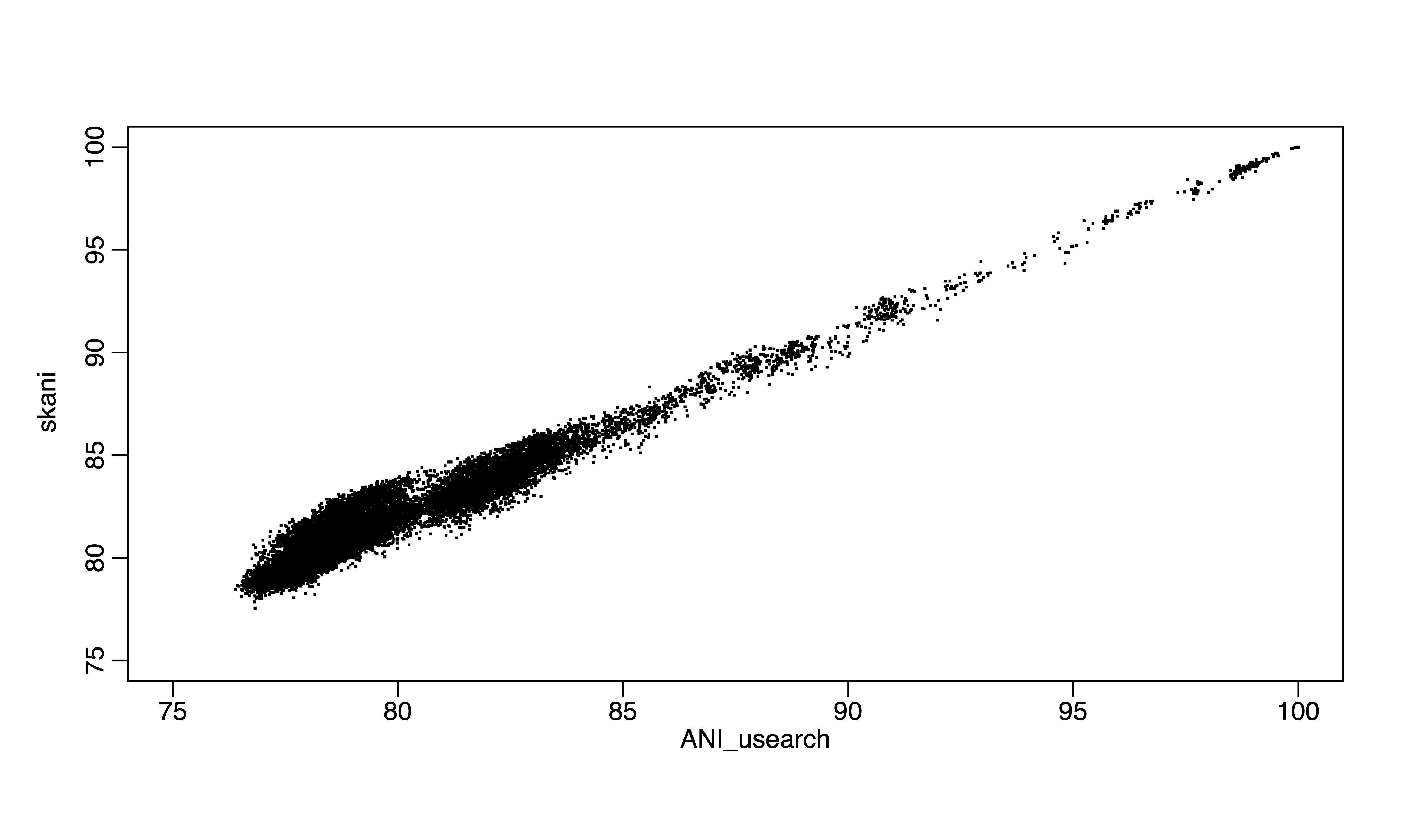 Figure 2. skani vs ANIu (usearch) for the same 300 genomes.
Figure 2. skani vs ANIu (usearch) for the same 300 genomes.
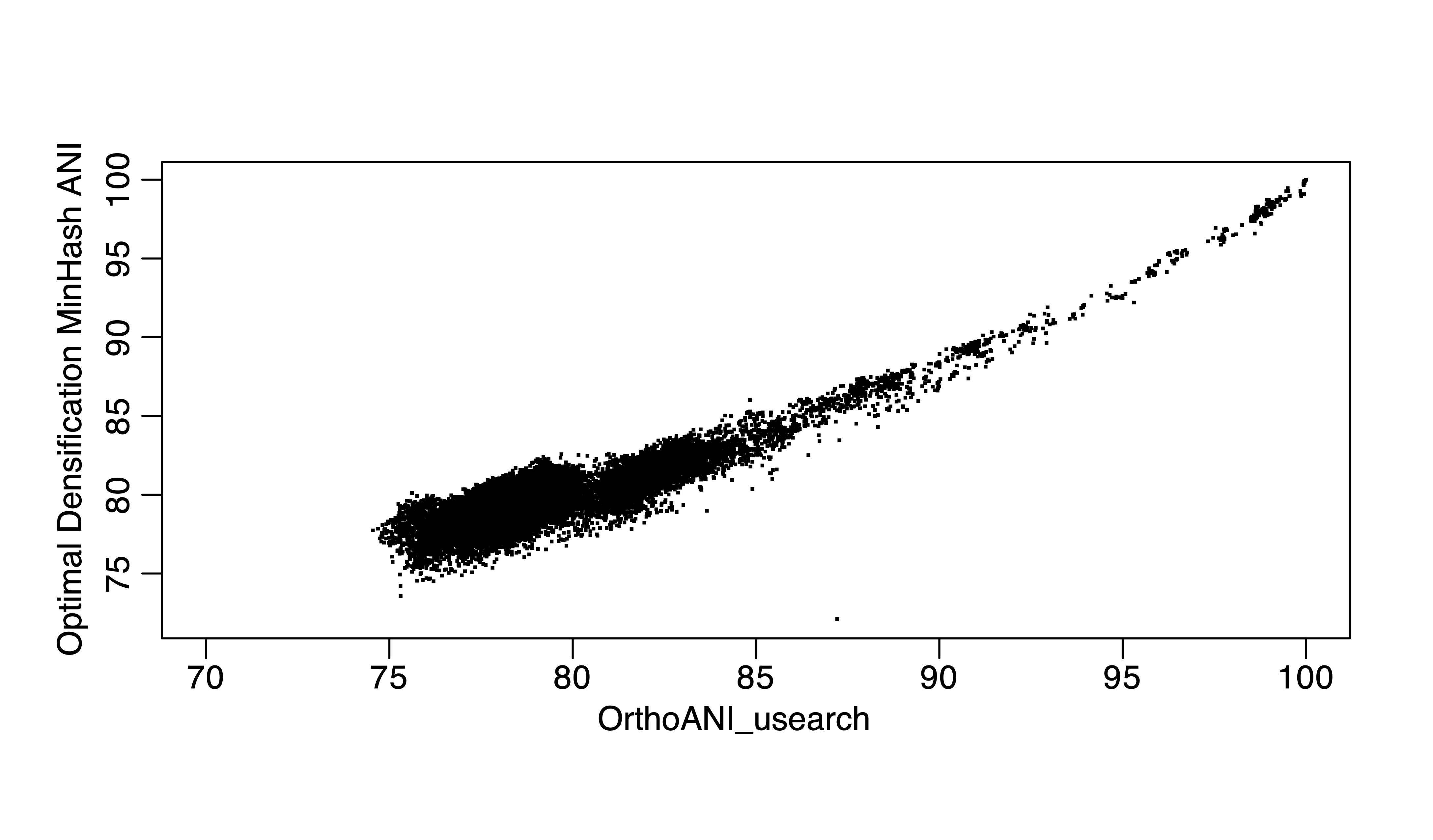 Figure 3. Optimal Densificatiin MinHash ANI vs ANIu for the same 300 genomes.
Figure 3. Optimal Densificatiin MinHash ANI vs ANIu for the same 300 genomes.
As you may notice, for below 80% ANI, fastANI is also less accurate (larger ANI values than ANIu) but with very small variation (standard deviation). It is very easy to apply a linear correction as shown, from the blue fitted line (fastANI vs ANIu) to red fitted line to accuractly approximate alignment-based ANI without any additional computations. Essentially, we want to learn a linear model (slope) after fixing (100,100) to minimize RMSE, which can be expressed as:
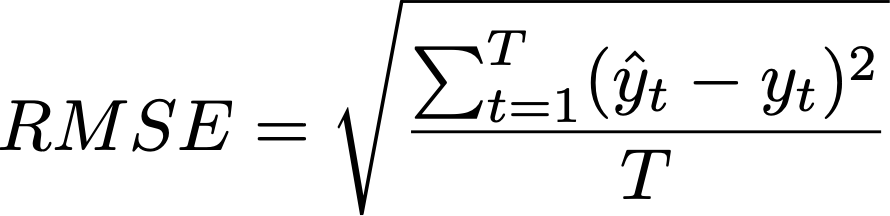 Equation 1. RMSE of FastANI using ANI_usearch as truth.
Equation 1. RMSE of FastANI using ANI_usearch as truth.
However, it is tedious to train such a linear model becasuse the all versus all alignment-based ANI for only several hundred genomes will take around a month or so on a single HPC node. We are now training a large-scale model with several thousand genomes (that is around 10 million ANI computations) using more than a thousand HPC nodes (each with a Intel(R) Xeon(R) Gold 6226 CPU).
![]() Figure 4. Linear correction of fastANI to approximate alignment-based ANI.
Figure 4. Linear correction of fastANI to approximate alignment-based ANI.
This has been applied in the newest version of fastANI (via the –correct option), which is available here. We will update the bioconda version soon.
fastANI -q query_genome.fasta -r reference_genome.fasta --correct -o output.txt
As expected, the corrected ANI value correlates perfectly with orthoANI:
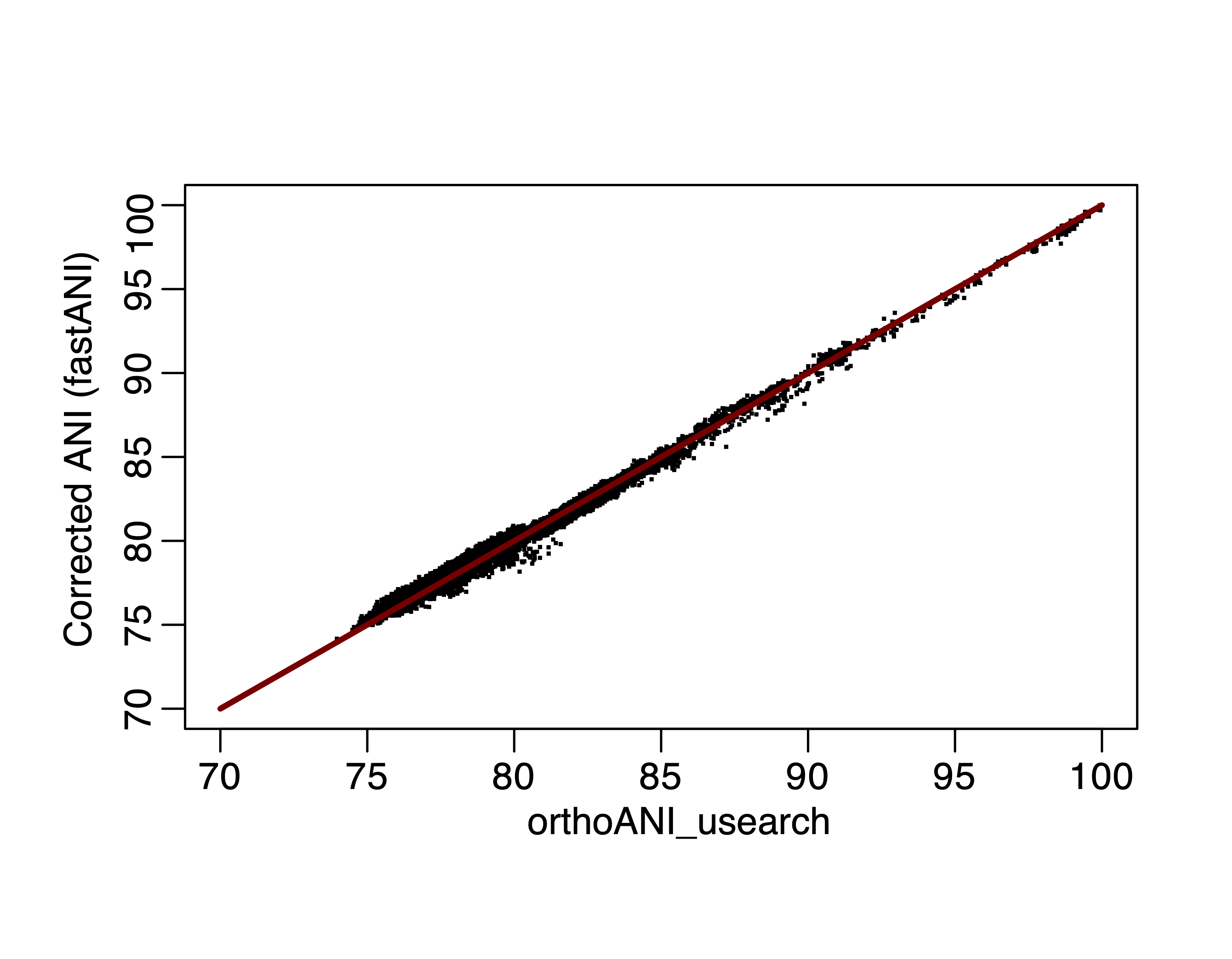 Figure 5. Corrected fastANI values versus orthoANI, fitting line is y=x.
Figure 5. Corrected fastANI values versus orthoANI, fitting line is y=x.
Based on all the analysis above, it is possible to have a new ANI calculator that is both fast and accurate, e.g. we use skani or optimal densification MinHash ANI for above 85% ANI estimation and fall back to corrected fastANI for below 85% ANI. We can almost approximate alignment-based ANI while being much faster.
References:
(1) Jain, C., Dilthey, A., Koren, S., Aluru, S., and Phillippy, A.M. (2017) A fast approximate algorithm for mapping long reads to large reference databases. In International Conference on Research in Computational Molecular Biology: Springer, pp. 66-81.
(2) Jain, C., Rodriguez-R, L.M., Phillippy, A.M., Konstantinidis, K.T., and Aluru, S. (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications 9: 5114.
(3) Shaw, J. and Yu, Y.W., 2023. Fast and robust metagenomic sequence comparison through sparse chaining with skani. bioRxiv, pp.2023-01.
(4) Shrivastava, A., 2017, July. Optimal densification for fast and accurate minwise hashing. In International Conference on Machine Learning (pp. 3154-3163). PMLR.